
累计签到:1361 天
连续签到:5 天
[LV.10]至尊爱粉
本帖最后由 子弹 于 2020-7-16 06:19 编辑
楼主可以关注一下 MET,药物如INC280 卡马替尼,还有BKM120,这个药物在-抗血管里面可以增效,小P在这两个药物里面联合的话。都很有效果。
抗-VEGF疗法通过c-Met抑制而复活
摘要:
一项新研究表明,通过使用抗VEGF抗体选择性抑制VEGF足以以c-Met依赖性方式增加侵袭和转移。抗VEGF治疗在神经内分泌胰腺癌的RIP-Tag2模型中诱导肿瘤缺氧,缺氧诱导因子1α和c-Met活化。选择性c-Met抑制足以阻断这些作用,为抗VEGF治疗提供了克服增加的侵袭的潜在机制和解决方案
介绍
VEGF诱导内皮细胞增殖,存活和迁移,并且是有效的血管通透因子。VEGF诱导的血管生成或来自现有血管网络的血管发芽在大多数成人组织中相对罕见,并且通常仅在伤口愈合和月经期间发生(1)。然而,血管生成是实体瘤生长所必需的,使其成为肿瘤治疗的有吸引力的靶标。许多目前的抗血管生成疗法靶向VEGF途径,VEGF途径是血管生成的主要驱动因素。

在2009年,作者在2篇文章强调了在临床前的多个肿瘤模型与阻断VEGF活性(策略增加的侵袭和转移治疗后3,4),抗VEGF治疗后的肿瘤表型的改变(5,6),并在这样做已设置边缘抗血管生成治疗领域。,在乳腺癌和黑色素瘤临床前模型中,针对VEGF途径的RTKi短期治疗后,转移加速并降低了存活率。在一项免费的研究中,通过使用各种策略(RTKi,单克隆抗体或遗传策略消除肿瘤细胞中的VEGF),报告在长期或短期VEGF抑制后神经内分泌胰腺癌(PNET)和胶质母细胞瘤模型中局部肿瘤细胞侵袭和远处转移增加)。

经提出了这些现象的几种可能的机制。肿瘤脉管系统的减少增加了肿瘤缺氧,可能选择更具侵袭性的肿瘤细胞群。此外,缺氧可能通过诱导或激活细胞外蛋白酶,诱导替代信号传导生长因子途径,或通过触发上皮 - 间质转化(EMT)样表型来刺激肿瘤细胞的侵袭(7)。此外,Ebos及其同事提供的数据(3)表明抗VEGF治疗可以调节远处器官,以允许肿瘤细胞外渗和转移性定植。现实情况是,这些提出的机制的组合可能是抗VEGF治疗后观察到的增加的侵袭和转移的原因。

研究结果
连续抗VEGF治疗可以减少原发肿瘤的生长并提高整体动物的存活率,但也可以增加肿瘤细胞在两种独立的胰腺癌模型中的侵袭和转移(8)证明了多RTKi(XL880),其抑制VEGFR和c-Met等,减少原发性肿瘤生长并阻止肿瘤细胞侵袭和转移。出抗VEGF治疗会增加肿瘤内缺氧,从而推动遗传程序导致c-Met表达和活性升高以及EMT介导的肿瘤细胞侵袭。以前,c-Met被认为是EMT和转移的驱动因素。

在本研究中,接受抗VEGF治疗的小鼠中肿瘤细胞的c-Met表达和活性显着增加,证实了缺氧与c-Met之间的已知关系。为了确定在抗VEGF治疗后观察到的增加的肿瘤侵袭和转移是否依赖于c-Met活性,在携带RIP-Tag2或Panc1原位胰腺肿瘤的小鼠中将c-Met抑制与抗VEGF治疗组合。联合治疗效果显着; 联合治疗完全消除了抗VEGF治疗后观察到的侵袭和转移的增加。事实上,联合治疗在RIP-Tag2模型中逆转了侵袭,治疗3周后,治疗开始时存在较少的瘤内淀粉酶阳性细胞。此外,在RIP-Tag2模型中,c-Met抑制显着降低了单独的肝转移并与VEGF抑制相结合,其中c-Met抑制与舒尼替尼的组合是最有效的。最后,作者使用双VEGFR2和c-Met RTKi(XL184)。在用XL184处理后,在RIP-Tag2模型中肿瘤细胞侵袭和转移显着减少,模仿先前当使用抗VEGF和c-Met靶向疗法的组合时所见的结果。

值得注意的是,尽管侵袭和转移增加,抗VEGF治疗单独延长了RIP-Tag2动物的存活,这反映了这些动物的原发肿瘤生长,侵袭,转移和发病率之间的总体平衡。然而,与单途径抑制相比,VEGF和c-Met信号传导的双重抑制提供了显着的存活益处。支持的整体模型是抗VEGF治疗修复血管系统并增加肿瘤内缺氧的模型。缺氧条件引发c-Met表达和活化增加,最终导致EMT样表型并增加肿瘤侵袭和转移。 |
|
|
任何一件不可能完成的事情,都是被分解成无数件可能完成小事情一个一个解决的。
|
|
|
|
 肺癌历经手术-复发-脑转移,如今母亲
讲述者:一只阿狸整理者:pear
熟悉我家的朋友都知道,我母亲虽然是IB期肺癌,但是很
肺癌历经手术-复发-脑转移,如今母亲
讲述者:一只阿狸整理者:pear
熟悉我家的朋友都知道,我母亲虽然是IB期肺癌,但是很
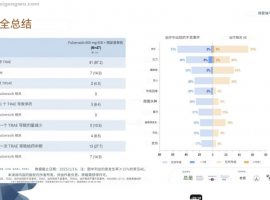 肺癌新疗法来袭!客观缓解率80%,双
作者:seacat
氟泽雷塞(商品名:达伯特®)是劲方医药开发的KRAS G12C 小分子抑制剂
肺癌新疗法来袭!客观缓解率80%,双
作者:seacat
氟泽雷塞(商品名:达伯特®)是劲方医药开发的KRAS G12C 小分子抑制剂
 急急急,求方案,希望得到大家的帮助
群里的老师们,请问如果肾转移会左侧腰以上痛吗?已经痛了有半个多月。一开始痛的时候
急急急,求方案,希望得到大家的帮助
群里的老师们,请问如果肾转移会左侧腰以上痛吗?已经痛了有半个多月。一开始痛的时候
 与myc结合亲和力高的已上市药物
MYC有所谓不可成药性,还没有专门的靶向药上市。
目前针对myc实际可行的治疗策略是根
与myc结合亲和力高的已上市药物
MYC有所谓不可成药性,还没有专门的靶向药上市。
目前针对myc实际可行的治疗策略是根
 67岁父亲肺腺癌4A期 L858R-伏美替尼
病情:
24年4月底我父亲因为呼吸不畅就医,抽胸水、做穿刺活检、做基因检测,结论
67岁父亲肺腺癌4A期 L858R-伏美替尼
病情:
24年4月底我父亲因为呼吸不畅就医,抽胸水、做穿刺活检、做基因检测,结论





 显身卡
显身卡



